
Kulturhaltung
Kultur von Reticulomyxa filosa
Im Rahmen dieser Arbeit wurden zwei Stämme von Reticulomyxa filosa eingesetzt, die beide von Hülsmann (1984) isoliert wurden. Eine Kultur stammt aus einem der Fischbecken in den Tropenhäusern des Botanischen Gartens der Ruhr-Universität, die andere aus einem See in der Umgebung von Berlin. Im Verlauf der Versuche zeigten beide Stämme keine Unterschiede, so daß auf eine Kennzeichnung verzichtet wird.
Die Zellen wurden in Petrischalen mit Medium 1:1 (Hülsmann, 1983) gehalten und zweimal wöchentlich mit Weizenkeimen gefüttert, wobei die Organismen, die sich vom Boden abgelöst hatten und an der Wasseroberfläche schwammen, in neue Schalen umgesetzt wurden. Die Zellen wurden bei Raumtemperatur im Halbschatten kultiviert.
| Ionenkonzentration | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ca2+ | Cl- | Mg2+ | SO42- | Na+ | HCO3- | K+ | HPO42- | NO3- | SiO32- | NH4+ | Ca2+ | EDTA | Fe3- | Cu2+ | Zn2+ | Co2+ | MO4- | CrO42- | VO2+ | Borat |
| 0,25 mM |
1,29 mM |
0,20 mM |
0,13 mM |
0,34 mM |
0,56 mM | 0,49 mM |
0,07 mM | 0,41 mM |
0,58 mM |
0,05 mM |
0,01 mM |
5,86 µM | 5,83 µM | 20,0 µM | 0,06 µM | 0,02 µM | 0,01 µM | 0,01 µM | 0,01 µM | 0,24 µM |
Tabelle 1: Zusammensetzung des Mediums 1:1Unter Berücksichtigung des Mineralgehaltes des verwendeten Tafelwassers (Volvic) ergeben sich die genannten Molaritäten für die wichtigsten Salze und Spurenelemente Der pH wurde nicht eingestellt, liegt aber normalerweise zwischen 6,8 und 7,2. |
||||||||||||||||||||
Experimentelle Bedingungen
Die Zellen wurden vor Beginn der Experimente in Gefäße mit frischem Kulturmedium überführt. Im Regelfall erhielten die Zellen dann 16 Stunden Gelegenheit, ein neues RPN auszubilden, bevor der Versuch begann. Für einige Versuche wurde die Auslaufzeit auf 2 - 4 Stunden verkürzt.
Für lichtmikroskopische Untersuchungen des RPN wurden die Zellen auf Deckgläsern kultiviert; die elektronenmikroskopischen Untersuchungen erfolgten zumeist an Organismen, die auf Thermanox-Plättchen oder ACLAR wuchsen.
Für die Elektrophorese aktiver Zellen wurden frisch angesetzte Schalen verwendet; für die Elektrophorese von Cystenproteinen wurden Zellen für 2 - 3 Wochen ohne Futter gehalten. Nach Ablauf dieser Zeit hatten sich die meisten Zellen in Cysten umgewandelt.
Da die Cysten bei einem Austausch des Kulturmediums oder anderen Milieuveränderungen häufig wieder ein RPN ausbilden, wurden zur Untersuchung der Cystenbildung einige Kulturen über mehrere Wochen in Hungerkultur gehalten. Mehrmals wöchentlich wurden in diesen Schalen alle neugebildeten Cysten mit einer Datumsmarkierung versehen. Nach einem Monat wurde dann der ganze Schaleninhalt fixiert.
Agenzien
Die Ausbildung des reticulopodialen Netzwerkes wurde in den Experimenten durch verschiedene Agenzien beeinflußt:
Herstellung von Zellmodellen
Durch die Behandlung von Zellen mit Detergenzien wurden Zellmodelle hergestellt, die mit verschiedenen Agenzien behandelt werden konnten. Je nach Versuchsziel wurde eines der beiden folgenden Protokolle verwendet:
Triton-Lysis im Kulturmedium
Das Kulturmedium wurde für 1 Minute gegen Medium 1:1 mit 1% Triton ausgetauscht und anschließend zweimal ausgewaschen. Diese Behandlung beeinflußt die Stabilität des Cytoskeletts nicht und wurde deshalb in Versuchen, in denen die Stabilität des Cytoskeletts gegenüber Pharmaka getestet wurde, eingesetzt.
Lysis in Vanadat-haltigem MT-stabilisierendem Medium
Das Kulturmedium wurde für 30 Sekunden bis 2 Minuten gegen LysismediumVIII ausgetauscht. Dieses Lysismedium wurde ursprünglich von Schliwa et al. (1991) zur Herstellung von stabilisierten Zellmodellen verwendet, an denen der Organellentransport reaktiviert wurde. Sein besonderer Vorteil liegt nicht nur darin, daß es Hexylenglykol als MT-stabilisierende Substanz enthält, sondern auch darin, daß das zugesetzte Vanadat die Querbrücken zwischen den Mikrotubuli in einem rigor-ähnlichen Zustand erhält. Dadurch werden die MT eines Bündels fest miteinander gekoppelt und gegen mechanische Beanspruchungen widerstandsfähiger gemacht.
- Lysismedium VIII
30,0 mM PIPES
12,5 mM HEPES
4,0 mM EGTA
1,0 mM MgCl2 pH 7,0 einstellen
0,15 % Brij 58
5,0 % Hexylenglykol
1,0 mM Vanadat zufügen
Das Lysismedium hält sich bei Raumtemperatur wochenlang; gelegentlich fiel jedoch das Detergens aus und mußte durch vorsichtiges Erwärmen auf 60-70 0C wieder gelöst werden.
Lichtmikroskopie
Lebenduntersuchungen
Lebenduntersuchungen wurden entweder mit den Photomikroskopen II oder III von Zeiss oder mit dem Zeiss-Umkehrmikroskop IM 35 ausgeführt. Makroskopische Beobachtungen wurden mit der Stereolupe M10 von Wild gemacht. Fotografiert wurde mit dem Negativmaterial TMax 100 oder TMax 400 von Kodak.
Präparate für die Umkehrmikroskopie wurden aus durchbohrten Objektträgern hergestellt, deren Öffnung auf der Unterseite mit einem Deckglas verschlossen und mit Silikonpaste (Baysilon) versiegelt wurde. Für die konventionelle Mikroskopie wurden Inkubationskammern aus einem Objektträger und einem Deckglas, das auf zwei Baysilon-Streifen lag, hergestellt. Diese Kammern erlaubten einen schnellen und vollständigen Austausch des Mediums. Für Langzeitbeobachtungen wurden die Objektträger in eine feuchte Kammer eingestellt.
Reflexionskontrast
Der Reflexionskontrast ermöglicht die Darstellung der Abstände zwischen Zellen und ihrem Untergrund, die im nm-Bereich liegen (Bereiter-Hahn & Vesely, 1994). Insbesondere kann man mit dem Reflexionskontrast die Anheftungsstellen von Zellen an dem Substrat sichtbar machen (Schindl et al., 1995). Für die Untersuchungen wurde das Umkehrmikroskop IM 35 mit einer Einrichtung zur Beobachtung im Reflexionskontrast, bestehend aus dem Objektiv Antiflex-Neofluar 63/1,25 (Öl) sowie einer Auflicht-Quecksilberleuchte, Polarisator und Analysator ausgerüstet.
Fluoreszenzmarkierungen
Phalloidin
Verschiedene Fixiergemische zur Darstellung des F-Actins, u.a. nach Czaban & Forer (1992) und Boyles et al. (1996) wurden erprobt. Am erfolgreichsten war jedoch das folgende Protokoll, das dann generell verwendet wurde:
Actin-Darstellung mit Rhodamin-konjugiertem Phalloidin:
- Lösung 1:
100,0 mM PIPES
25,0 mM HEPES
10,0 mM MgCl2 pH 7,0 einstellen,
10 % DMSO zufügen und auf Endvolumen auffüllen - Lösung 2:
- 100,0 mM PIPES
25,0 mM HEPES
10,0 mM MgCl2
20,0 mM Spermidinphosphat pH 7,0 einstellen,
10 % DMSO zufügen und auffüllen.
Unmittelbar vor Gebrauch wurde 7%para-Formaldehyd durch Erhitzen auf 700C in Lösung1 gelöst, danach abfiltriert und wieder auf Raumtemperatur abgekühlt. Zum Fixieren werden gleiche Teile beider Lösungen gemischt und unmittelbar auf die Zellen gegeben. Nach 30-minütiger Fixierung wurde das Gemisch dreimal mit Ca-freier PBS ausgewaschen.
- Ca-freie PBS
8,0 g/l NaCl
1,15 g/l Na2HPO4 * 2 H2O
0,2 g/l KH2PO4
0,1 g/l MgCl2 * 6 H2O pH 7,2
Die Phalloidinmarkierung erfolgte mit Rhodamin-konjugiertem Phalloidin (30 Minuten), das aus einer 3,3 µM Stammlösung in Methanol 1:20 mit Caf-PBS verdünnt wurde. Danach wurden erneut dreimal mit Caf-PBS gespült und das Präparat in Einbettungspuffer überführt.
- Zusammensetzung des Einbettungspuffers
90,0 ml Glycerin (87%)
0,1 g para-Phenylendiamin
9,0 ml Ca-freie PBS pH 8,5 mit NaHCO3 einstellen
Beobachtung im Reflexionskontrast mit anschließender Actin-Darstellung
Um die Anheftungsstellen der Zellen im Reflexionskontrast mit dem Vorkommen von F-Actin vergleichen zu können, wurden die Zellen im Reflexionskontrast beobachtet und fotografiert. Unmittelbar danach wurden die Zellen fixiert. Um das Wiederauffinden des interessierenden Bereichs zu erleichtern, wurde eine weitere Fotografie nach der Fixierung angefertigt und die Objektstelle zusätzlich mit einem Objektmarker eingekreist.
Anschließend wurde eine Actin-Darstellung mit Phalloidin durchgeführt, und die fotografierten Bereiche des RPN wurden anhand von Markierung und Fotografie identifiziert.
Indirekte Immunfluoreszenz
Um die räumliche Verteilung verschiedener Tubulinmodifikationen oder von MTOCs darstellen zu können, wurden folgende Antikörper eingesetzt:
| AK | Typus | Ursprungs- organismus | Antigen | Referenzen | Spezifität | Verdünnung | |
|---|---|---|---|---|---|---|---|
| IIF | WB | ||||||
| YL 1/2 | IgG | Ratte | Hefe-Tubulin | (Kilmartin et al., 1982; Wehland et al., 1983; Wehland et al., 1984) | Tyr-a-Tubulin | 1:10 | 1:5 |
| 27C2 | IgM | Maus | Tyr-Peptid | (Wehland & Weber, 1987) | Tyr-a-Tubulin | 1:10 | 1:10 |
| 20C6 | IgG1 | Maus | Tyr-Peptid | (Wehland & Weber, 1987) | Tyr-a-Tubulin | 1:10 | 1:10 |
| ID5 | IgG1 | Maus | Glu-Peptid | (Wehland & Weber, 1987) | Glu-a-Tubulin | 1:10 | 1:5 |
| CTR 210 | IgM | Maus | humane Lymphoblasten Centrosome | (Bailley et al., 1988) | Centrosom | 1:50 | 1:50 |
| hVIN1 | IgG1 | Maus | humanes Vinculin aus dem Uterus | (Goncharova, 1992) | Vinculin | 1:200 | - |
| TAT1 | IgG2b | Maus | a-Tubulin | (Woods et al., 1989) | a-Tubulin | 1:5 | 1:5 |
| Tub 2-28-33 | IgG1 | Maus | Seeigel b-Tubulin | (Siddiqui, 1989) | b-Tubulin | 1:1000 | 1:1000 |
| YOL 1/34 | IgG2a | Ratte | Hefe-Tubulin | (Kilmartin et al., 1982) | a-Tubulin | 1:1000 | 1:500 |
| Nr. 11 | ? | Maus | b-Tubulin | (Koonce et al., 1986) | b-Tubulin | 1:100 | 1:50 |
| WA3 | ? | Maus | Rinderhirn-b-Tubulin | - | b-Tubulin | 1:10 | 1:10 |
| anti- Reti-culomyxag-Tubulin | polyklonal | Kaninchen | Reticulomyxa-g-Tu-
bulin |
(Kube-Granderath & Schliwa, 1996) | g-Tubulin | 1:100 | 1:100 |
| anti
g-Tubulin |
polyklonal | Kaninchen | synthetisches Oligopeptid aus konservativer g-Tubulin-Sequenz | (Joshi et al., 1992) | g-Tubulin | 1:200 | - |
| 6-IIB-1 | IgG | Maus | acetyliertes a-Tubulin | (Piperno & Fuller, 1985) | acetyliertes a-Tubulin | 1:10 | 1:10 |
| anti-Actin | ? | Kaninchen | Actin | - | Actin | 1:5 | - |
| anti-
Centrin |
? | Maus | Centrin von Spermatozopsis similis | (Lechtreck et al., 1989) | Centrin | 1:10 | - |
Tabelle 2: Verwendete Erstantikörper |
|||||||
Fixierung und Antikörperinkubation für die IIF
Ähnlich wie bei der verwandten Foraminifere Allogromia laticollaris (Golz, 1986) lassen sich die MT von Reticulomyxa filosa mit den herkömmlichen Azeton- oder Methanolfixierungen sowie mit Formaldehyd in situ nicht adäquat fixieren. Stattdessen wurde in Anlehnung an Koonce & Schliwa (1986) mit 1% Glutaraldehyd + 0,1% Triton in Caf-PBS fixiert. Im Anschluß wurden die Zellen dreimal in Caf-PBS gespült und für 20 Minuten mit 1 mg/ml NaBH4 in einem Gemisch aus gleichen Teilen Caf-PBS und Ethanol behandelt, um die fixierungsbedingte Autofluoreszenz zu unterdrücken. Das Borhydrid wurde in drei Waschgängen in 1% BSA in Caf-PBS ausgespült, dann wurde der Erstantikörper mit Caf-PBS + 1% BSA verdünnt aufgetragen, für mindestens eine Stunde bei 370C inkubiert und dreimal ausgewaschen.
| AK | Ursprungs- organismus | gegen | Konjugat | Adsorbiert gegen | Verdünnung |
|---|---|---|---|---|---|
| Dianova 115-015-062 | Ziege | Maus | DTAF | Mensch, Rind, Pferd | 1:100 |
| Dianova 115-015-100 | Ziege | Maus | DTAF | Ratte, Mensch, Rind, Pferd | 1:100 |
| Dianova 715-015-150 | Esel | Maus | DTAF | Huhn, Ziege, Meerschwein, Hamster, Kaninchen, Schaf, Mensch, Rind, Pferd | 1:100 |
| Dianova 115-025-071 | Ziege | Maus IgG Fcg Fragment | TRITC | Mensch, Rind, Pferd | 1:100 |
| Dianova 115-015-075 | Ziege | Maus IgM µ-Kette | DTAF | Mensch, Rind, Pferd | 1:100 |
| Dianova 111-025-144 | Ziege | Kaninchen | TRITC | Mensch, Maus, Ratte | 1:100 |
| Dianova 112-015-062 | Ziege | Ratte | DTAF | Mensch, Rind, Pferd | 1:100 |
| Dianova 712-025-153 | Esel | Ratte | TRITC | Huhn, Ziege, Meerschwein, Hamster, Maus, Kaninchen, Schaf, Mensch, Rind, Pferd | 1:100 |
Tabelle 3: Verwendete Zweitantikörper |
|||||
Der Zweitantikörper wurde ebenfalls für mindestens eine Stunde in Caf-PBS + 1% BSA bei 370C inkubiert und im Anschluß gründlich mit Caf-PBS ausgewaschen. Anschließend erfolgte die Überführung in Einbettungspuffer.
Heterologe Tubulin-Polymerisation
Um Tubulin an bestehende MT oder MTOCs von Reticulomyxa anzupolymerisieren, wurden die Zellen für eine Minute in Lysismedium VIII lysiert und dann mit PM-Puffer (Simon & Salmon, 1990) mit 1 mM GTP und wechselnden Konzentrationen an Hirntubulin bei 370C für 30 Minuten inkubiert. Anschließend wurde mit Glutaraldehyd oder Methanol fixiert und wie ein normales Immunfluoreszenzpräparat weiterbehandelt.
- PM-Puffer
100,0 mM PIPES
2,0 mM EGTA
1,0 mM MgSO4 pH 6,9 einstellen
Das anpolymerisierte Hirntubulin wurde mit dem mAB YL 1/2 nachgewiesen, da dieser AK mit Reticulomyxa-Tubulin nicht reagiert (siehe Ergebnisteil).
Auswertung der fluoreszenzmarkierten Präparate
Die Auswertung der Präparate erfolgte mit einem Zeiss PhotomikroskopII, das mit einer Auflicht-Fluoreszenzeinrichtung ausgerüstet war. Die Fotos wurden mit den Objektiven Planapo 63/1,4 (Öl) oder Planapo 40/1,0 (Öl) und dem Film Kodak TMax400 gemacht.
| Fluoreszenzfarbstoff | DTAF | FITC | Rhodamin | TRITC |
|---|---|---|---|---|
| Anregungswellenlänge | 450 - 490 nm | 450 - 490 nm | 546 nm | 546 nm |
Tabelle 4: Fluoreszenzfarbstoffe und Anregungswellenlängen |
||||
Elektronenmikroskopie
Epon-Einbettungen und Ultradünnschnittechnik
Fixierung
Die Zellen wurden mit 2,5% Glutaraldehyd in 0,1M Cacodylatpuffer pH7,0 für 45 Minuten fixiert und anschließend dreimal mit Cacodylatpuffer gespült. Zur selektiven Kontrastierung von Mikrotubuli durch Tannin wurde bei einigen Versuchen dem Fixiermedium 4% Tannin aus einer 8% igen Stammlösung in Aqua bidest zugegeben.
In allen Fällen wurde nach der GA-Fixierung mehrmals gespült und für 15 Minuten mit 1%OsO4 in 0,1M Cacodylatpuffer pH7,0 auf Eis nachfixiert.
Entwässerung und Epon-Einbettung
Die Entwässerung und Einbettung der Zellen erfolgte nach der Methode von Luft (1961), wurde jedoch hinsichtlich der Austauschzeiten, der Zusammensetzung des Einbettungmittels und der Polymerisationstemperatur verändert.
Die Zellen wurden über eine aufsteigende Ethanolreihe von 30%, 50%, 70%, 90% und 96% (je 10 Minuten), sowie 100% (zweimal 10 Minuten) in reines Propylenoxid überführt. Das Propylenoxid wurde einmal ausgewechselt und die Objekte wurden über die Konzentrationsreihe
- Epon / Propylenoxid 1/3 30 Minuten
- Epon / Propylenoxid 1/1 1 Stunde
- Epon / Propylenoxid 3/1 1,5 Stunden
in reines Epon überführt. In diesem verblieben sie über Nacht, bevor die Polymerisation durch Erwärmung auf 600C für drei Tage durchgeführt wurde.
- Epon-Mischung (hart)
48,21 ml Glycidether
16,66 ml DDSA
34,60 ml MNA
2,0 ml DMP 30 Das fertig angesetzte Epon wird portioniert eingefroren und gelagert.
Schnittherstellung und -auswertung
Nach der Aushärtung des Epons wurden die interessierenden Bereiche des RPN ausgesägt und in der gewünschten Orientierung auf Blöckchen aufgeklebt, die dann mit einem Reichert OM U2 oder Reichert Ultracut E Ultramikrotom in ca. 70 nm dicke Schnitte zerlegt wurden.
Die Schnitte wurden auf Formvar-beschichteten Kupfergrids aufgefangen und wie üblich mit 2% Uranylacetat in Aqua bidest und Bleicitrat nach Reynolds (1963) kontrastiert.
Die Schnitte wurden in einem Philips EM 410 bei 80 kV ausgewertet und auf dem orthochromatischem Film EG 18 von Guilleminot dokumentiert.
"Hook-Dekoration"
Wenn Mikrotubuli unter geeigneten Bedingungen mit einem Tubulin-Überschuß inkubiert werden, erfolgt nicht nur eine Verlängerung der MT, sondern das Tubulin polymerisiert auch blättchenförmig an bestehende Mikrotubuli an. Im Querschnitt erscheinen die blattförmigen Polymerisate als hakenförmig gebogen. Die Drehrichtung dieser Häkchen kann verwendet werden, um die Polarität von Mikrotubuli zu bestimmen. Dabei weist nach der "Rechte-Hand-Regel" (Heidemann & McIntosh, 1980) der Daumen in Richtung des minus-Endes der MT, wenn die Finger die gleiche Krümmung wie die Tubulin-Hooks aufweisen.
Tubulin-Isolation für die "Hook-Dekoration"
Das Hirntubulin für die "Hook-Dekoration" wurde zumeist frisch aus Schweinehirn nach der Methode von Tiwari & Suprenant (1993) isoliert. Wenn Tubulin nicht unmittelbar zur Verwendung kam, wurde es tropfenweise in flüssigem Stickstoff eingefroren und darin bis zum Gebrauch gelagert.
Durchführung der "Hook-Dekoration"
Die "Hook-Dekoration" bei Reticulomyxa wurde nach einer modifizierten Methode von Heidemann (1991) durchgeführt. Die Zellen liefen auf ACLAR- oder Thermanox-Plättchen aus, wurden danach eine Minute mit Lysismedium VIII behandelt und zweimal für wenige Sekunden mit Dekorationsstammlösung gespült, die mit Aqua bidest auf die Hälfte verdünnt war.
- Dekorationsstammlösung:
1,0 M PIPES
2,0 mM EGTA pH mit konzentrierter NaOH-Plätzchen auf 6.9 einstellen
2,0 mM MgCl2
5,0 % DMSO
Im Anschluß wurden die Zellen wurden für 20 Minuten bei 370C mit Dekorationspufferlösung inkubiert, mit Glutaraldehyd fixiert und simultan tanninkontrastiert. Die Zellen wurden eingebettet und senkrecht zu den Pseudopodien geschnitten.
- Dekorationspufferlösung (ansetzen und lagern auf Eis)
500,0 µl Dekorations-Stamm
1,0 mM GTP
1,5 mg Tubulin
ad 1 ml mit eiskaltem Aqua bidest
Dieses Protokoll wurde in verschiedenen Versuchen durch Variation von Inkubationstemperatur (250C bis 370C), EGTA-Gehalt (1 bis 8 mM), DMSO-Konzentration (2,5 - 10%) und Tubulinkonzentration (1 - 2 mg/ml) verändert, erwies sich aber in der aufgeführten Form für Reticulomyxa optimal.
Auswertung der "Hook-Dekoration"
Von jedem ausgewählten Bereich des reticulopodialen Netzwerkes wurde eine Schnittserie angefertigt. Auf einer Skizze der Schnittfläche wurde jedes Mikrotubulibündel vermerkt, um Doppelzählungen zu vermeiden. Um die Drehrichtung aller Häkchen eines MT-Bündels sicher bestimmen zu können, mußten die Schnitte mit einem Goniometerhalter im EM so lange gekippt werden, bis senkrecht auf die jeweiligen MT geblickt werden konnte. Die Mikrotubulibündel wurden bei geringer MT-Anzahl direkt im EM oder sonst auf Fotos von Kippserien ausgezählt.
Bei einem Teil der Mikrotubulibündel wurde zusätzlich zu Anzahl und Drehrichtung der Häkchen auch die Gesamtanzahl der MT sowie der Aggregate aus MT und anpolymerisiertem Tubulin bestimmt.
Es wurde große Sorgfalt darauf verwandt, alle möglichen Drehungen des Objektes während der Präparation und der Ultradünnschnitte im EM zu protokollieren, um hinsichtlich der Polarität der MT korrekte Ergebnisse zu gewährleisten.
Die Bewertung der Auswertbarkeit von Tubulin-Häkchen sowie ihrer Drehrichtung erfolgte wie bei Heidemann (1991).
Fig. 3: Interpretation der Drehrichtung von Tubulin-HooksAlle Bilder auf der rechten Seite sind als Häkchen im Uhrzeigersinn (C) oder gegen den Uhrzeigersinn (C') interpretierbar. Die Bilder auf der linken Seite werden als unauswertbar erachtet. Die Häkchen im Uhrzeigersinn erkennt man an Mikrotubuli, wenn man auf das langsamwachsende oder minus-Ende sieht. (Verändert, nach Heidemann (1991)) |
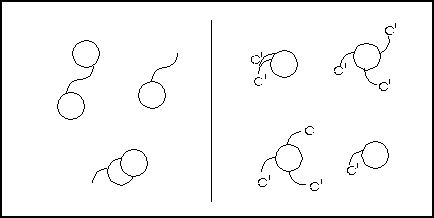 |
Gelelektrophorese
SDS-PAGE
Die SDS-PAGE wurde nach der Methode von Laemmli (1970) durchgeführt. Die verwendeten Gele waren entweder 10- oder 12,5%-Gele mit einem Acrylamid/Bisacrylamid Verhältnis von 30/0,8. In Abhängigkeit vom Verwendungszweck wurden für den Western Blot Gele von 100 x 80 x 0,4 mm, für Standardzwecke 180 x 160 x 0,8 mm oder in der 2. Dimension der 2D-PAGE 180 x 160 x 1,6 mm eingesetzt.
Wenn eine Bestimmung des Proteingehaltes von Proben als notwendig erachtet wurde, erfolgte die Proteinbestimmung nach Schaffner & Weissmann (1973) mit Amidoschwarz, da diese Meßmethode die genauesten Ergebnisse liefert und zudem nicht gegen den Elektrophorese-Probenpuffer empfindlich ist (Henkel & Bieger, 1994).
Die Gele wurden mit 12% TCA in Aqua bidest fixiert und mit 0,1% Coomassie-Blau in 50% Methanol und 10% Essigsäure gefärbt. Überschüssiger Farbstoff wurde in 50% Methanol + 10% Essigsäure ausgewaschen. Anschließend wurden die Gele mit Agfaortho O25 fotografiert und nach der Methode von Wiedbrauk et al. (1991) getrocknet.
Molekulargewichtsbestimmung
Zur Bestimmung der Molekulargewichte in der Elektrophorese wurde der Molekulargewichtsmarker SDS-6-H verwendet. Zur Berechnung der Gewichte unbekannter Proteine wurde das Programm PROTMW (Duggleby, 1994) eingesetzt.
| Protein | Myosin | b-Galactosidase | Phosphorylase B | BSA | Ovalbumin | Carbonic Anhydrase |
|---|---|---|---|---|---|---|
| Molekulargewicht | 205 KDa | 116 KDa | 97,5 KDa | 66 KDa | 45 KDa | 29 KDa |
Tabelle 5: Zusammensetzung des Molekulargewichtsmarkers |
||||||
Western Blot
Der WB der 100 x 80 x 0,4 mm-Gele wurde als Semi-Dry Blot nach der Methode von Khyse-Andersen (1984) durchgeführt. Gele von 1,6 mm Dicke wurden entweder für 1,5 Stunden geblottet, oder der Stromfluß wurde für eine Stunde auf 1,2 mA / cm2 erhöht.
Mit Ausnahme der Proben für das MALDI-TOF (s.u.) wurde stets auf gewebeverstärkte Nitrocellulose von 0,2 µm Porengröße geblottet. Die Blots wurden entsprechend den Standardprozeduren blockiert und mit Antikörpern inkubiert. Es fanden die gleichen Erstantikörper Verwendung wie in der IIF (S. 17).
Die Zweitantikörper waren Meerrettich-Peroxidase konjugierte polyklonale Antikörper von Ziegen gegen Maus-, Ratten- oder Kaninchenimmunglobuline, die in einer Verdünnung von 1:1000 eingesetzt wurden. Die Detektion der gebundenen Zweitantikörper erfolgte mit 4-Chloronapthol und H2O2 als Substrat.
2-Dimensionale Elektrophorese
Die 2D-PAGE erfolgte nach der Methode von O'Farrell (1975), modifiziert nach Hochstrasser et al. (1988). Die isoelektrische Fokussierung erfolgte in 200 µl-Kapillaren; die zweite Dimension lief auf 180 x 160 x 1,6 mm großen Gelen. Im Anschluß an die zweite Dimension wurden die Gele entweder wie von Hochstrasser et al. (1988) silbergefärbt, oder der Kernbereich der Gele wurde ausgestochen und dem WB unterzogen.
Bestimmung der isoelektrischen Punkte
Die isoelektrischen Punkte wichtiger Proteine wurden durch Mischung der Proben mit dem IEF-Marker "Protein Test mixture 9" (SERVA 39206) und Kolokalisation der Proteine in der Silberfärbung interpoliert.
| Protein | Cyto- chrom C | Ribonuclease A | Lectin | Myoglo- bin | Carboan- hydrase | b-Lacto- globulin | Trypsin Inhibitor | Glucose- oxidase | Amyloglu- cosidase |
|---|---|---|---|---|---|---|---|---|---|
| MW | 12.384 | 13.700 | 49.000 | 17.000 | 29.000 | 18.400 | 20.100 | ? | 70.000 89.000 |
| IEP | 10,65 | 9,45 | 7,75 / 8,0 / 8,3 | 6,9 / 7,35 | 6 | 5,15 / 5,3 | 4,5 | 4,2 | 3,5 |
Tabelle 6: Zusammensetzung des IEP-Markers |
|||||||||
Massenspektroskopie
Zur Präzisierung der Molekulargewichtsbestimmung wurde von ausgewählten Proteinen das Molekulargewicht mit Hilfe der MALDI-TOF Massenspektroskopie bestimmt. Bei dieser Technik werden die Proteine zuerst in der Gelelektrophorese aufgetrennt und auf eine Membran geblottet. Die geblotteten Proteine werden dann in eine Matrix aus kleinen, hochabsorbierenden organischen Molekülen eingebettet.
Durch kurzzeitigen Beschuß mit hochenergetischem Laserlicht entstehen aus den Proteinen Ionen, deren Masse in einem Time-Of-Flight Massenspektrometer bestimmt wird.
Fig. 4: Lokalisation der Proben für die MassenspektroskopieDie Proteine wurden auf PVDF-Membranen geblottet und die Membran in drei Längsstreifen unterteilt. Auf dem rechten Längsstreifen wurde die Position des untersuchten Proteins mit monoklonalen Antikörpern bestimmt; der linke Längsstreifen wurde mit Coomassie-Blau gefärbt. Der mittlere Streifen wurde im MALDI-TOF Massenspektrometer in Höhe der Antikörpermarkierung gemessen. |
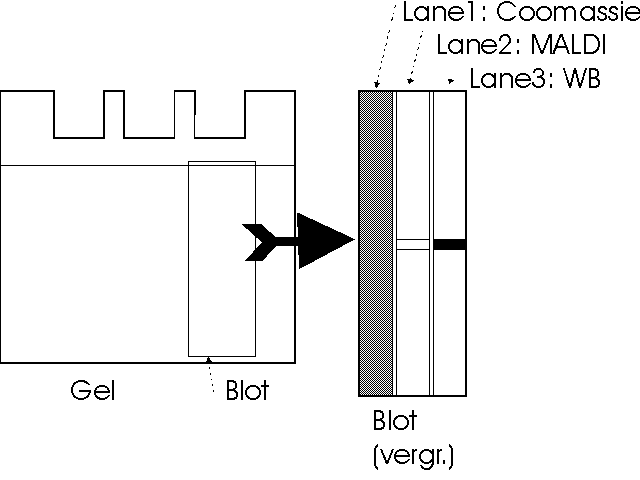 |
Die Proben für die Massenspektroskopie wurden nach Strupat et al. (1994) vorbereitet: Nach der SDS-PAGE wurden die Gele auf PVDF-Membranen geblottet und die Streifen wurden der Länge nach gedrittelt. Das linke Drittel wurde mit Coomassie-Blau entsprechend den Standardprozeduren gefärbt. Das rechte Drittel wurde mit Antikörpern inkubiert, um die präzise Lage der gesuchten Proteine auf dem Blot zu finden. Der mitlere Abschnitt der Streifen wurde mit der Matrixlösung (40g/l Bernsteinsäure) inkubiert, getrocknet und in Höhe der Antikörpermarkierung auf dem rechten Abschnitt mit 80 ns Impulsen aus einem Infrarot-Laser angeregt. Die freigesetzten Ionen wurden im Massenspektrometer gemessen. Je nach Güte der Spektren wurden zwischen 11 und 22 Messungen summiert. Als Blindwert wurde eine mit Proteinen unbeladene Stelle der Membran gemessen und von dem Meßwert der Proteine abgezogen.
Die Messungen im Massenspektrometer wurden unter Mitarbeit von Frau Dr. Kerstin Strupat (Universität Münster) durchgeführt; Messungen für die Vorversuche wurden von Frau Dipl. Phys. Jianru Zeng (Universität Münster) und Frau Dipl. Phys. Kathrin Breuker (ETH Zürich) durchgeführt.
Bezugsquellennachweis
Antikörper
Die Antikörper YL 1/2, 27C2, 20C6 und ID5 wurden freundlicherweise von Prof. Dr. Wehland bereitgestellt. Der Antikörper gegen Reticulomyxa-g-Tubulin stammte von Herrn Dr. Kube-Granderath. Der Antikörper gegen g-Tubulin wurde uns von Herrn Joshi zugesandt. Der Antikörper CTR 210 wurde von Herrn Bornens zur Verfügung gestellt. Herr Gull war so freundlich, mir eine Charge des Antikörpers TAT1 zuzusenden. Frau Dr. Euteneuer schulden wir Dank für die Antikörper WA3 und Nr. 11. Der Antikörper 6-IIB-1 stammte von Dr. Fuller. Der Antikörper gegen das Spermatozopsis-Centrin wurde uns von Herrn Dr. Melkonian zur Verfügung gestellt. Der Antikörper YOL 1/34 stammte von der Firma Calbiochem (Bad Soden/Ts); die Antikörper hVIN1 und Tub 2-28-33 wurden bei Sigma (Deisenhofen) erworben.
Die Peroxidase-konjugierten Zweitantikörper wurden von DAKO (Hamburg) erworben.
Chemikalien
- ACLAR Plano, Marburg
- ATP Boehringer, Mannheim
- Baysilon Bayer, Leverkusen
- DDSA Serva, Heidelberg
- DMSO Baker, Groß Gerau
- DMP 30 Serva, Heidelberg
- DNP Fluka, Neu Ulm
- EGTA Serva, Heidelberg
- Glycidether Serva, Heidelberg
- GTP Sigma, Deisenhofen
- HEPES Serva, Heidelberg
- Hexylenglykol Serva, Heidelberg
- MgCl2 Riedel-de-Haën, Hannover
- MNA Serva, Heidelberg
- Natriumorthovanadat Sigma, Deisenhofen
- Nitrocellulose Sartorius, Göttingen
- Nocodazol Sigma, Deisenhofen
- Nonidet Fluka, Neu Ulm
- Osmiumtetroxid Degussa, Hanau
- PIPES Sigma, Deisenhofen
- Propylenoxid Serva, Heidelberg
- Rhodamin-Phalloidin Sigma, Deisenhofen
- SDS-6-H Sigma, Deisenhofen
- Taxol NIH, USA
- Thermanox Miles Lab., Inc
- Vinblastinsulfat (Velbe) Lilly Research Lab., Greenfield, IN
Alle nicht aufgeführten Chemikalien wurden von der Fa. Merck, Darmstadt bezogen